マルチパスIn-Situスペクトルイメージング法を用いた分散露光
はじめに
時間経過や外部刺激に対する試料の変化を捉える能力はその場分析の重要なポイントです。従来、その場観察条件における変化を電子顕微鏡で捉える手法は主に像観察でした。それに加え、電子エネルギー損失分光法(EELS)のスペクトルイメージングのような分光法を基にした観察方法が増加してきています。しかしながら、従来の光学的に結合されたCCDやCMOSセンサーの読み出しノイズが微小ではあっても、化学的に敏感であり同時に微弱なコアロスの現象の観察に対してはその場EELSスペクトルイメージング(EELS-SI)法の適用が困難でした。主にポアソン統計によってのみ制限される直接検出器のEELSへの採用によって[1]、コアロスエッジを用いたIn-Situ EELS-SIが実現可能となり、化学結合と化学反応の僅かな変化を高い空間分解能と時間分解能で観察・分析するための扉を開きます。加えて、分割露光を行ったEELS-SIはマルチパスデータ取得におけるスタンダードなモードとして用いることが可能であり、試料ドリフトの補正、データ取得後の利用不可能なパスの排除、本来の状態を維持した試料から取得したパスのみ積算する、といったことが実現します。
本アプリケーションノートでは、電子エネルギー吸収端微細構造(ELNES)を用いて炭酸カルシウム試料に電子線を照射した際の化学変化を観察し、その還元機構を考察しています。
実験結果
市販の炭酸カルシウム(CaCO3)は、還元反応および質量損失に対する総照射電子線量のしきい値が十分に評価されています[2,3]。炭酸カルシウムナノ粒子は蒸留水に分散させ、厚さ25 nmのSiN支持膜上に滴下、乾燥しました。STEMスペクトルイメージデータは日本電子株式会社製JEM-F200を使用して加速電圧200 kV、ビーム電流96 pAで取得しました。また、スペクトルは、ピクセルドウェルタイムが0.004 sec/pixel(ローロススペクトルは0.001秒、ハイロススペクトルは0.003秒)でDualEELS機能を使用し取得しています。ピクセルサイズは1 nmでさらに4×4のサブピクセルスキャンを行っており、隣接するサブピクセル間でビームが重なり合うことを防いでいます(ビーム径は2 Å以下です)。サブピクセルスキャンは、ピクセル中央だけではなくピクセル全体に照射範囲を拡げます。GatanのDigitalMicrograph® 3.6に新たに搭載されたIn-Situ SI機能を使用することで、個々のパスを独立して保存することが可能です。エネルギードリフトの補正は、全てのSIデータに対してデータ取得後に行うことも可能ですが、本測定ではZLP-Lock機能を用いてライブで行いました。空間のドリフトのアライメントについても、連続的に走査しながらのライブドリフト補正と取得後のドリフト補正が可能です。全てのEELSのスペクトルイメージは室温(25℃)で取得しています。
CaCO3 粒子の投影面積は総照射電子線量に応じて減少しており、その変化を図1aに示します。また、その形態変化も総照射電子線量の増加と共に観察され、ADF像の挿入図からも判るようにCaCO3 粒子は端部にファセットを有する立方体のような形状から端部が丸まった直方体のような形状へと変化します。図1bの粒子の相対厚さも照射電子線量の増加と共に減少していることを示しています。図1aの投影面積の減少と併せて考えると、体積、即ち質量損失がその成分の昇華(放射線分解)を通じて生じていることが確認出来ました。

DigitalMicrograph 3.6中のIn-Situツールを使用することで、炭酸カルシウム試料に対する電子線照射量の影響を考察するためにその場スペクトルイメージデータのアライメント、切り取り、積算操作を行うことが出来ます。CaCO3粒子の平均組成は、フルエンスの増加と共に変化することが先行する研究によって報告されています[2]。In-Situスペクトルイメージングの新しい機能とContinuum®の収集効率の向上、そしてK3® カメラの感度を組み合わせることによって、異なる総フルエンスにおける元素マップが取得可能となりました。結果として、形態の変化と局所的な組成の変化を相関付けることが出来ます。図2に明らかな形態変化が現れた際のフルエンスにおけるスペクトルイメージデータから抽出した結果を示します。元素マップはC K、O K、Ca L2,3 エッジから生成しています。面積密度の最も大きな変化(コントラストの低下)は、CとOのマップには観察されますが、一方でCaマップにおける総信号強度はほぼ一定です。図2aと2b中に青色矢印で示すADF像中のコントラストが暗い領域は、空間的にはC、O、Caの密度の低下に対応しています。ADF像と各マップ中の信号強度の低下は、恐らくボイドの不均一な核生成によるものであり、このボイドはフルエンスの増加と共に粒子内を伝播することが観察されています。このボイド形成の不均一さは、核生成のしきい値の低い欠陥に起因する可能性があります[2]。粒子の形態の変化は、元素マップにおいては不均一に生じることが観察されています。図2cと図2dにおいて、粒子の上方と左右に丸まった端部が形成されています。しかしながら、粒子の下方ではファセットが残っており、その部位を赤色矢印で示します。フルエンスの増加と共に、ADF像と元素マップの双方でファセットが粒子の中心に向かって拡がっています。粒子は総フルエンスが1.41 x 106 e-/Å2を超えると楕円形となります。以前の研究では、CaCO3からCaOへの還元は等方的に発生し、粒子の端部から始まり中心へと向かって伝播すると報告されていました[2]。しかしながら、図2の元素マップとADF像に示されるように、CaCO3の還元は異方性を示していました。図2eの最終的な元素マップでは、OとCの元素マップ上にCaの元素マップを重ね描きしています(マゼンタの破線)。Cの信号の投影面積は3046 nm2 (±116 nm2)であり、一方OとCaの信号はそれぞれ4019 nm2 (± 128 nm2)と4335 nm2 (± 130 nm2)です。これはCとOの損失を通じたCaCO3の放射線分解による還元を示唆しています。CaCO3が大気中で熱分解する際にCO2の生成が観察され、最終的な還元生成物はCaOとなります[3]。しかしながら、図2eに観られるように、炭素Cの投影面積はOの投影面積よりも30%小さくなっています。このことは放射線分解時にOと比較してCが高濃度で失われているか、あるいは残留炭酸塩相と共にCaOの多孔質のネットワークが形成されていることを示唆しています。後者は、CaCO3粒子の還元の際に空孔が形成されることをSTEM BF像で観察した研究[2]とよく一致しています。図2eのCの元素マップを詳細に評価すると、信号強度(密度)の僅かな変化が観察されており、残留したCに多孔質のネットワークが形成されていることを示唆しています。

図3に初期状態(図2a)と終状態(図2e)の元素マップから抽出した濃度プロファイルを示します。濃度プロファイルは、図3に挿入したADF像上に青色の矢印で示した向きで粒子から抽出しています。初期の元素マップでは、ストイキメトリが公称のCaCO3から外れています(図3a)。OとCaの比率は2.2:1(公称3:1)で、OとCの比率は3.2:1(公称3:1)です。この公称値との乖離は、一部は合成時のばらつきによるものであるが、主に電子線照射によるダメージよって生じたものであると考えられます。図3bの粒子の終状態から抽出した濃度プロファイルでは、OとCaの比が1:1に近い事を示しています。一方、O とCの比は5:1に増加しています。このCaの密度の増加とOの密度の減少は、放射線分解中にCaOの形成を観察した報告と合っています。粒子中央部のOの密度は粒子端部と比較して僅かに高くなっており、CaCO3のような相が残留し粒子が完全に還元していないことを示唆しています。

ELNESは、電子構造の局所的な変化を研究するために使用することが可能であり、その結合や結晶対称性がフルエンスによってどのように変化するかについての情報を明らかにします。図4の重ね描きしたプロットは、各フルエンスにおけるC K、Ca L2,3、O KエッジのELNESの変化を示しています。図4aのC Kエッジは、フルエンスの増加と共に最も少ない変化を示しています。ELNESの初期形状は炭酸塩の特徴を示しており、aからdの4つの異なるピークを示します[5,6]。289 eVの初めのシャープなピークは、CO32-アニオンの非占有π*軌道への遷移に由来しており、301 eVのブロードなピークはCO32-アニオンの非占有σ起動への遷移に由来します[6]。σ*ピーク上の肩のaとbはCO32-クラスター周りの占有殻からの散乱によるものと考えられています[6]。フルエンスの増加と共にσ*ピーク上の肩のaとbがはじめに消失し、CO32-アニオンの周りの局所的な対称性が減少したことを示唆しています。π*のピークは徐々に強度が減少し、一方σ*のピークはブロードになっています。しかしながら、π*とσ*の両方が粒子の最終状態において存在しており、これは粒子内でCとOがまだ互いに結合していることを示唆しています。2.38 x 105 e-/Å2までのフルエンスの増加後、非常に弱いπ*ピークライクな非晶質カーボンのπ̽ピーク(C-C結合)が観察されています。非晶質カーボン膜から得られたC-Kエッジをリファレンスとして上部にプロットしています。
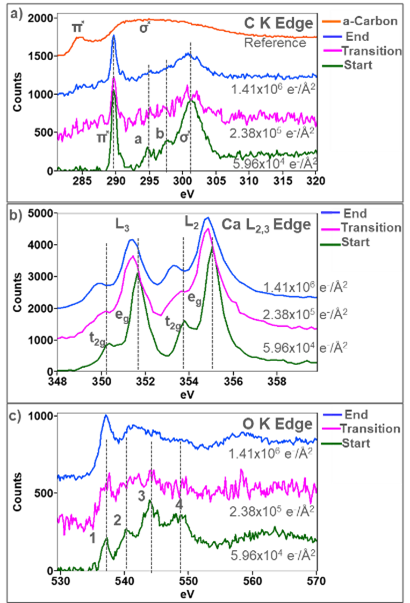
図4bでは、Ca L3エッジとL2エッジのegピークとt2gピークの間に1.26 eVの分裂が観察され、これは純粋なCaCO3試料から得られたCa L2,3と一致しています。総フルエンスが増加するにつれて、egとt2g間の分裂が減少しており、これは結晶場分裂エネルギーの変化を示しています。Ca L2,3エッジの結晶場分裂はCaイオン周りの局所的な対称性に対して非常に敏感です。RezとBlackwellは、結晶のCaCO3と非晶質のCaCO3の間でegとt2gの分裂に同様の減少が生じることを示しています[7]。フルエンスの増加と共に、L3ピークとL2ピークはより低いエネルギーへとシフトし、egとt2gの間の分裂は1.4 eVに増加することが観察され、これはCaOから得られた微細構造と一致しています[3, 8]。Golla-Schindleらは、CaCO3が結晶性CaOへと再結晶する前に非晶質/ディスオーダー相へと還元する同様の転移を観察しています[5].
図4cにおいて、O Kエッジは1から4とラベルを付けた4つの異なるELNESのピークを有しており、CaCO3について以前に報告されているO KのELNESの形状と一致しています[3]。ピーク1は、Oの2p軌道とCaの3d軌道の混成に起因しています[4,9]。このピークはフルエンスが増加しても存在しており、CaCO3からCaOへの遷移を通じてCaが常にOに結合していることを示唆しています。またピーク2から4は、Oの2pのS軌道との混成によるものと考えられます。この範囲のO Kエッジの形状は、O 2p状態とCa 4sおよびC 2s状態との混成によって決まります[9]。フルエンスが増加すると、ピーク2から4は消失し、これはCaの還元とCO32-アニオンからの炭素の離脱を示唆しています。O Kエッジの最終的な形状は、過去にCaOで観察されたELNESの形状と類似しています[3]。しかしながら、CaOから取得したO KのELNESでは、第三のピークが観察されており図4c中の最終的なスペクトルとはなっていません。最終的なスペクトルに明確な第三のピークが存在しないことは、CaO相は形成されたが多数のOの空孔が存在していることを示唆しています。図4に示したELNESの解析では、ELNESのフィンガープリンティングがフルエンスの増加に伴ってどのように変化するかの平均を決定するため、意図的に粒子全体から合算したスペクトルを使用して行っています。しかしながら、K3カメラの優れた感度によって、はるかに少ない数のスペクトルからELNESの解析を行うことが出来ます。従って、相マップも高い空間分解能で生成され、粒子全体に対して局所的な微細構造の変化を調べることが可能となりました。
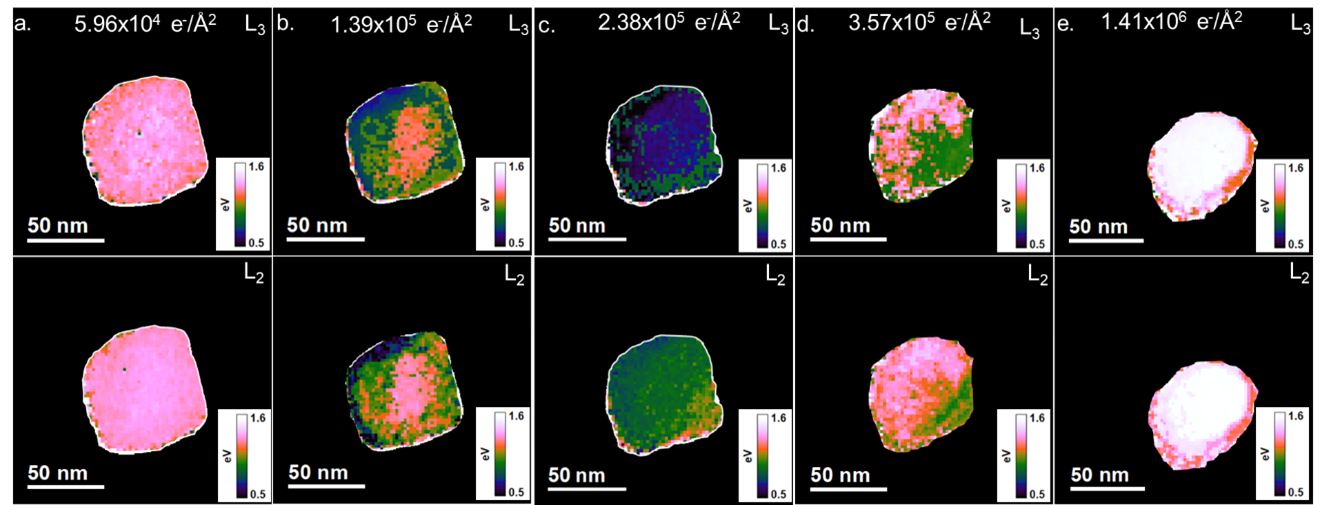
図5に、L3とL2エッジのegとt2gピークの間の分裂を粒子上の位置に対してプロットしました( E(eg)-E(t¬2g))。ピーク位置は、DigitalMicrographソフトウェアのLeast square(LLS)ツールを使用し、L3とL2エッジ内の各ピークに対してガウス関数を用いて非線形最小二乗フィッティングを使用し決定しています。初期は最も大きな分裂が粒子の中央部で観察され、端部に近づくにつれて減少しています。総フルエンスが増加するにつれてeg-t2gの分裂に勾配が生じます。eg-t2gの分裂は、図1と図2のADF像で観察されていた右下のファセットが観察されていた部分と比較して、丸みを帯びた端部が観察された領域である粒子の上方と左方では小さくなっています。ファセットの消失と共に、分裂は粒子の右下で減少し上方と左方で増加しています。最終状態では再び粒子中央部で最大の分裂が発生しており、L3とL2ピークのeg-t2g分裂は端部に向かって減少します。これはCaCO3からCaOへの中間ディスオーダー相への遷移が粒子内で不均一に起こることを示唆しています。このような微細構造の局所的な変化は、図4bのCa L2,3エッジのELNESの平均的な変化を解析するだけでは観察することが出来ません。

図6 O Kエッジ(左列)とC Kエッジ(右列)のMLLSフィッティングを用いた相マップ。図4中にプロットしたELNESを内部リファレンスとして使用。
DigitalMicrographソフトウェアの多変量線形最小二乗法(multiple linear least squares、MLLS) のスタンダードをベースとした定量ツールを用いて、図6に示すCとOの相マップを生成しました。各粒子の試料厚さの違いを考慮するため、多重散乱の補正を有効にしています。図4にプロットしたELNESを、C KとO KのELNESの内部スタンダードとして利用しました。図6の一列目では、O KのELNESはフルエンスの増加と共に炭酸塩相から酸化カルシウム相への遷移を示しています。初期状態では、粒子の端部にディスオーダー相が観察され、総フルエンスが高くくなると共に大きなボイドが発生し始めます。遷移状態では、粒子に主にディスオーダー相が現れ酸化カルシウム相も僅かに観察されます。最終的に観察される状態では、主に酸化カルシウム相が観察されています。
図6の二列目、C KエッジのELNESではさらに小さな遷移を示しています。初期状態では、炭酸塩相のみが存在しています。フルエンスが増加するにつれて、非晶質相がより多く観察されています。しかしながら炭酸塩相は完全には消失していません。炭酸塩相のELNESに対するより強いフィッティングは、図4cの粒子の最終状態から抽出したELNESで観察された強いπ*とσ*ピークの存在に依るものと考えられ、CO32-アニオンがまだ粒子内に存在することを示唆しています。 しかしながら、それらがどのように新たなCaO相に取り込まれているのかは明らかになっていません。
まとめ
In-situ マルチパススペクトルイメージングとK3カメラの優れた感度を組み合わせることで、これまでの研究では達成することが出来なかった空間分解能で放射線分解中のCaCO3粒子の変化のキャラクタリゼーションが可能となりました。投影面積と試料厚さはフルエンスの増加と共に減少し、放射線分解による質量の減少を示唆しています。CaCO3粒子中のC、Ca、Oの形態変化と濃度(面積密度)の減少は、フルエンスの増加と共に観察されています。組成の最も大きな変化は、CとOの損失に起因するものであり、これらは恐らくはCO2へと変化し真空中で昇華したと思われます。K3カメラの優れた感度によって、C K、Ca L2,3、O KエッジのELNESの解析を通じてCaCO3からCaOへの相転移を確認することが出来ました。またその高い空間分解能によって、CaCO3の還元は粒子の端から中央に向かって等方的にではなく異方的に起こっていることが観察されました。さらにCa L2,3エッジの分裂を解析することで、これまでの報告と一致する中間的なディスオーダー相が観察されています[5]。CaOが形成された後でも、CO32-アニオンが粒子中に存在することが観察されています。これは、CaOが形成されるとCは完全にCO2へと変換されることと示唆したこれまでの報告とは異なっています[5]。還元されたCaCO3粒子が多孔質構造を形成したことは、TEM明視野像とSTEMで示されています。これらのこれまでの報告と本研究で観察された結果に鑑みると、粒子の最終的な状態は残存する炭酸塩相と混在するCaO結晶の多孔性ネットワークである可能性が高いと考えられます。
参考文献
[1] J. Hart et. al., Sci Rep. 7 (2017) 8243 doi : 10.1038/s41598-017-07709-4.
[2] R. Hooley, A. Brown, R. Brydson Micron 120 (2019) p25-34 doi: 10.1016/j.micron.2019.01.011.
[3] AWD Hills, Chemical Engineering Sciences 23 (1968) pg297-320 doi: 10.1016/0009-2509(68)87002-2.
[4] R. F. Egerton et. al., Ultramicroscopy 110 (2010) 991-997 doi : 10.1016/j.ultramic.2009.11.003
[5] U. Golla-Schindler, G. Benner, A. Orchowski, U. Kaiser, Microsc. Microanal. 20 (2014) pg715-722 doi: 10.1017/S1431927614000464.
[6] L. A .J. Garvie, A.J. Craven, and R. Brydson, R. Am Mineralogist (1994) 79, pg411–425.
[7] P. Rez, A. Blackwell, J. Phys. Chem. B 115 (2011) 11193-11198.
[8] F. Hofer, P. Golob, Ultramicroscopy 21 (1987) p379-383 doi: 10.1016/0304-3991(87)90036-2
[9] F. M. F. de Groot et. Al. Phys. Rev. B. 40 (1989) 5715 doi: 10.1103/PhysRevB.40.5715.